The haem synthetic pathway
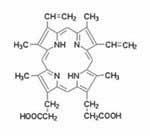
The porphyrias are classified as metabolic disorders, arising from defects in the biosynthetic pathway leading to the production of haem (haem synthetic pathway). In this pathway, glycine and succinyl-CoA combine to form the first porphyrin precursor, aminolaevulinic acid (ALA); two molecules of ALA then combine to form the second porphyrin precursor porphobilinogen (PBG), and four molecules of PBG combine to form uroporphyrinogen. Uroporphyrinogen is the first true porphyrin, showing the characteristic tetrapyrrole configuration of a porphyrin. (Click on the diagram at left for a description of the tetrapyrroles). Uroporphyrinogen then undergoes a systematic rearrangement of its hydrocarbon side chains, resulting in a series of different porphyrinogens as shown in the pathway in the figure below . The penultimate compound is protoporphyrin: insertion of an atom of iron results in the compound haem.
Porphyrins and porphyrinogens
The tetrapyrroles exist in two forms: a reduced form known as the porphyrinogens and an oxidised form known as the porphyrins. The porphyrinogens (uroporphyrinogen, coproporphyrinogen etc.) constitute the intermediates on the haem synthetic pathway, but tend to become oxidised when they escape from this pathway into blood, urine and stool: it is therefore the porphyrins which are measured in the laboratory.
Enzymes of the haem synthetic pathway

Haem is required in erythroid tissue for the production of hemoglobin, as well as in non-erythroid tissues (principally the liver), where it is incorporated into many haemoproteins, including the cytochromes. This allows a division of the porphyrias into two major categories: the erythropoietic porphyrias, in which the enzyme defect manifests in the haem synthetic pathway within erythroblasts, resulting in the accumulation of porphyrin within red blood cells, and the non-erythropoietic orhepatic porphyrias, in which porphyrins are overproduced in the liver.
The enzyme defect
in most cases the defective enzyme is caused by a mutation in the gene coding for that enzyme. Most porphyrias are therefore genetic disorders, and may be transmitted as autosomal dominant or recessive conditions. This is described in more detail in the accompanying pageInheritance of porphyria. An important exception is porphyria cutanea tarda (PCT), which is usually an acquired, rather than an inherited, disorder.
Clinical Effects of Porphyria
These are twofold: the acute attack and photocutaneous sensitivity. The acute attack, also known as the acute porphyric crisis, is marked by severe abdominal pain and an autonomic neuropathy (typically presenting as hypertension, tachycardia and ileus), which may progress to a motor neuropathy with respiratory failure and death. Photocutaneous sensitivity typically presents with increased skin fragility, blistering, erosions and scars in sun-exposed areas. Each porphyria varies in its propensity to cause acute attacks, skin disease or both.
The clinical effects and mode of inheritance are summarized in the following table. Further information on each porphyria is available from the menu.
| CATEGORY |
TYPE
|
CLINICAL PRESENTATION
|
INHERITANCE
|
| Hepatic | ALA dehydratase deficiency | Acute attacks | Autosomal recessive |
| Acute intermittent porphyria | Acute attacks | Autosomal dominant | |
| Porphyria cutanea tarda | Skin disease | Usually acquired; a minority are inherited (autosomal dominant) | |
| Hereditary coproporphyria | Skin disease, acute attacks | Autosomal dominant | |
| Variegate porphyria | Skin disease, acute attacks | Autosomal dominant | |
| Erythropoietic | Congenital erythropoietic porphyria | Skin disease | Autosomal recessive |
| Erythropoietic protoporphyria | Skin disease: specific presentation with immediate photosensitivity | Autosomal dominant: severe forms have complex inheritance |